Відповідно до чинного законодавства України невід’ємною частиною процесу реєстрації, перереєстрації та забезпечення імпорту лікарських засобів на територію України є надання документа, що підтверджує умови виробництва лікарських засобів вимогам GMP:
- Висновок про визнання GMP сертифікатів, виданих уповноваженими органами країн – членів ЄС, Великої Британії або країн, що мають угоду про взаємне визнання з ЄС або з Україною
або
- Сертифікат відповідності умов виробництва лікарських засобів вимогам GMP.
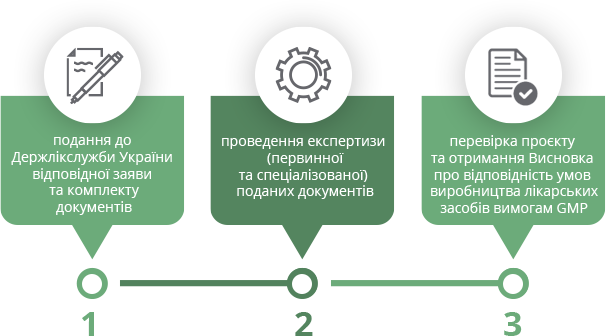
Видання висновків або сертифікатів здійснюється центральним органом виконавчої влади – Державною службою України з лікарських засобів та контролю за наркотиками (далі – Держлікслужба України) – відповідно до Порядку проведення підтвердження відповідності умов виробництва лікарських засобів вимогам належної виробничої практики, затвердженого наказом МОЗ України від 27.12.2012 № 1130 (далі – Порядок) та розробленого з урахуванням вимог Директиви 2001/83/ЄС Європейського Парламенту.
Підтвердження відповідності умов виробництва лікарських засобів чинним в Україні вимогам GMP запроваджено з метою доведення, що лікарські засоби постійно виробляються і контролюються згідно зі стандартами якості, які відповідають їхньому призначенню, а також відповідно до вимог реєстраційного досьє, досьє досліджуваного лікарського засобу для клінічних випробувань або специфікації на цю продукцію.